
Recently we published a blog on cutting through the jargon of medical device development, which proved to be hugely popular. As a follow-up to that blog, we thought we would share some insights on the many regulatory standards that must be adhered to in order to ensure the safety and usability of a medical device throughout its development.
In the medical device world, the numbers associated with these standards are used colloquially and for a newcomer to the industry, this can be quite confusing. So, as a ‘beginner’s guide’, what are the development standards that are important to consider when developing a medical device?
The Standards
ISO 13485 – Quality Management
ISO 13485 specifies the requirements for a Quality Management System (QMS) where an organisation needs to design, produce or maintain medical devices and related services.
A company whose QMS is certified to this standard will have been audited to ensure that the relevant processes and procedures are in place, so that medical devices can be developed, manufactured and deployed safely, with the full technical development and document audit history. Certified companies are audited regularly to ensure that adherence to their processes and procedures is maintained.
ISO 14971 – Risk Management
Risk management is an important factor in the development of medical devices. It ensures that potential risks of using a medical device have been considered and mitigated against.
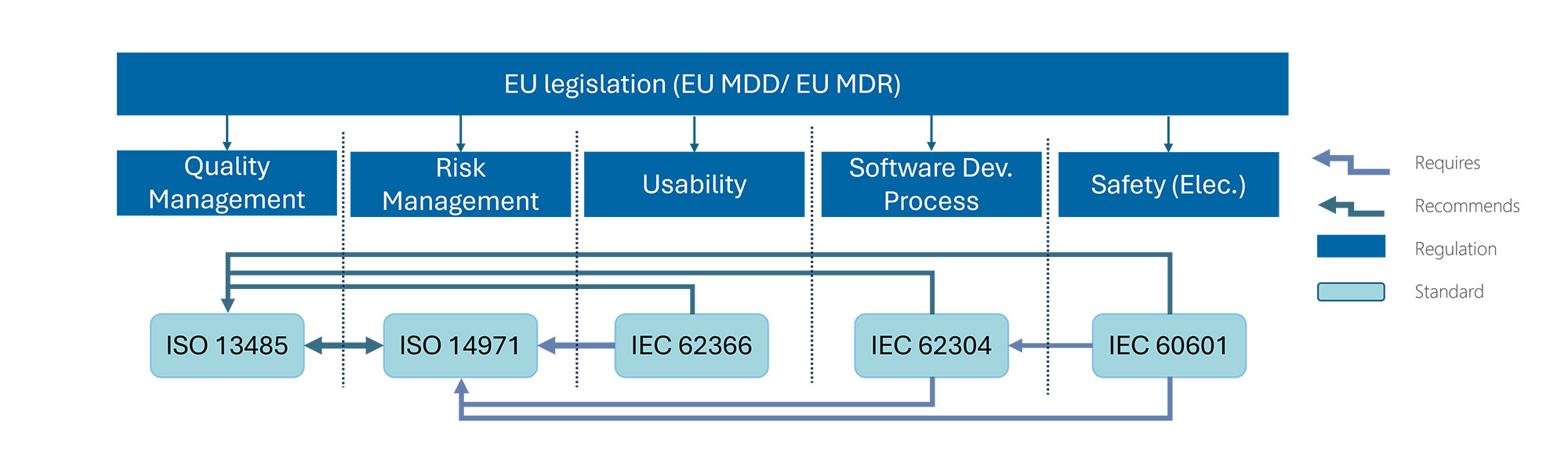
ISO 14971 specifies the process of risk management surrounding medical devices, software as a medical device (SaMD) and in-vitro diagnostic (IVD) devices. It is referred to by ISO 13485, IEC 62366, IEC 62304 and IEC 60601 as the standard to follow for risk management. Management of risk is also a significant component of the applicable regulations including MDR, IVDR and FDA requirements
It allows designers and manufacturers of medical devices to identify the associated hazards, estimate and evaluate the associated risks, control these risks, and monitor the effectiveness of those controls.
IEC 62304 – Software Development
Software for medical devices must be developed to IEC 62304, which encompasses the processes and procedures required and establishes a common framework for the medical device software lifecycle process.
The IEC 62304 software standard ensures that software development encompasses requirements capture, risk analysis, architectural and detailed design, code implementation, validation, verification and traceability throughout the entire development process. It also covers the update, configuration and maintenance processes required after software is released in a device.
There is a corresponding standard, IEC 82304, which applies to software that is developed for use on general computing platforms, such as desktop computers and mobile devices.
IEC 61010 – Electrical Safety (IVDs)
IEC 61010 provides general safety guidelines and requirements for the development of all electrical equipment for measurement, control and laboratory use meaning it is the key electrical safety standard applicable to In-Vitro Diagnostic Devices (IVDs) The standard is extensive and includes a general section dedicated to safety, along with many sub-sections detailing explicit requirements to deal with safety associated with different types of electrical equipment.
IEC 62366 – Usability Engineering for Medical Devices
IEC 62366 is the international standard that describes the application of usability engineering to medical devices. By applying human factors to the design, development and testing of a medical device, it allows all aspects of device/human interaction to be considered and a safe and effective device to be developed. The human factors process may involve early-stage user research and usability testing, as well as analysis of use-related issues and risk to elicit the way that people will interact with a product. This will inform the design making the end product easier to use.
IEC 60601 – Basic Safety and Performance Considerations (Medical Devices)
IEC 60601 covers the general and essential performance requirements for all medical electrical equipment. Although often referred to as electrical safety and EMC the 60601 family of standards actually covers a much more extensive range of requirements. This includes specific aspects of safety and performance in areas such as electrical emissions and immunity. In addition to this, the standard addresses human safety, so that it can be proved that the medical device can be used without harming the operator or the patient.
How do the standards fit together?
As well as having an appreciation of the applicable standards, it is also important to understand that hierarchy in which these standards exist. ISO 13485 is the overarching standard in which all medical device development takes place. ISO 14971 for the management of risk is also referenced by the other medical device development standards.
Regulatory bodies
It is impossible to just talk about the standards without having some discussion on the regulatory bodies that oversee the medical device approval process.
There are many national bodies that regulate the MedTech market, but the major ones are described below:
- United States: Food and Drug Administration (FDA)
- European Union: European Commission (EC)
- United Kingdom: Medicines and Healthcare products Regulatory Agency (MHRA)
- Canada: Health Canada (HC)
- Japan: Pharmaceutical and Medical Devices Agency (PMDA)
- Australia: Therapeutic Goods Administration (TGA)
- China: National Medical Products Administration (NMPA)
- India: Central Drugs Standard Control Organization (CDSCO).
- FDA
The USA’s Food and Drug Administration group manages food safety, drug safety and the regulation of medical devices.
- EC – European Community
The European Medicines agency has overall responsibility for the regulatory process, but individual EU states regulate at the state level.
- MHRA
The MHRA is responsible for regulating the medical devices market in the UK. All medical devices, including in vitro diagnostic devices, need to be registered with the MHRA before they are placed on the market. It is worth noting that Northern Ireland has certain exemptions and a UKCA mark or an EU CE mark can be applied for.
Conclusion
Hopefully, this article has given a good overview of the standards, what they cover and how they all fit together.
eg technology are ISO 13485 accredited, meaning we put rigorous quality management at the centre of everything we do, providing our clients with the compliance to standards for safety, quality, risk management and traceability. We keep abreast of any regulatory developments in the MedTech sector, ensuring every medical device we develop is carried out to the latest standards.
If you would like to find out more about what medical device standards would apply to your product development, please get in touch and we will be more than happy to discuss your project and how we optimise your route to market.
If would like to chat with one of our team about your product design and development requirements, please do not hesitate to get in touch:
Via email on design@egtechnology.co.uk, by giving us a call on +44 01223 813184, or by clicking here.



